Neuartige KI-basierte Software ermöglicht schnelle und zuverlässige Abbildung von Proteinen in Zellen
Software entwickelt, die Proteine in der Kryo-Elektronentomographie genau erkennt, auswählt und so die mühsame händische Selektion ersetzt

Die Kryo-Elektronentomographie (Kryo-ET) entwickelt sich zu einer leistungsfähigen Technik zur Abbildung detaillierter 3D-Bilder von zellulären Umgebungen und eingeschlossenen Biomolekülen. Eine der Herausforderungen bei dieser Methode ist jedoch die Identifizierung von Proteinmolekülen in den Bildern für die anschließende Prozessierung am Computer. Ein Forscherteam um Stefan Raunser, Direktor am MPI für molekulare Physiologie in Dortmund, hat unter der Leitung von Thorsten Wagner eine Software entwickelt, um Proteine in überfüllten zellulären Räumen zu identifizieren. Das neue Open-Source-Tool mit dem Namen TomoTwin nutzt metrisches Deep Learning basierend auf neuronalen Netzen, und ermöglicht es Forschenden, mehrere Proteine mit hoher Genauigkeit und hohem Durchsatz zu lokalisieren, ohne das neuronale Netz jedes Mal manuell neu erstellen oder neu trainieren zu müssen.
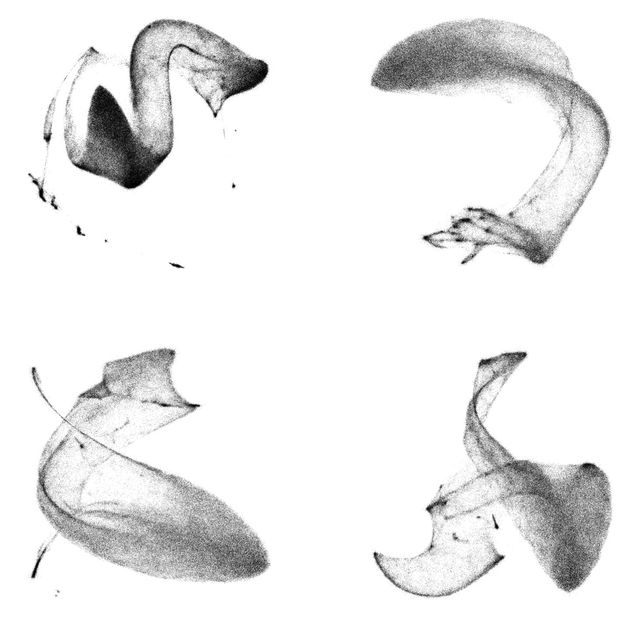
TomoTwin-Verarbeitungskarten für ein auf 2D abgeflachtes Tomogramm. Partikel verschiedener Makromoleküle werden in der Karte entsprechend ihrer Struktur angeordnet, so können verschiedene Makromoleküle in Zellen identifiziert und lokalisiert werden.
MPI of Molecular Physiology

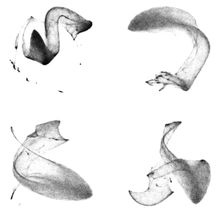
Erstautoren Gavin Rice (links) und Thorsten Wagner (rechts).
MPI of Molecular Physiology

Je mehr, desto besser
"TomoTwin ebnet den Weg für die automatisierte Identifizierung und Lokalisierung von Proteinen direkt in ihrer zellulären Umgebung und erweitert damit das Potenzial der Kryo-ET", sagt Gavin Rice, Co-Autor der Studie. Die Kryo-ET hat das Potenzial, die Funktionsweise von Biomolekülen in einer Zelle zu entschlüsseln und damit die Grundlagen des Lebens und die Entstehung von Krankheiten aufzudecken.
Bei einem Kryo-ET-Experiment erstellen die Forschenden in einem Transmissions-Elektronenmikroskop 3D-Bilder, so genannte Tomogramme, eines zellulären Raums mitsamt den darin enthaltenen komplexen Biomolekülen. Für ein detaillierteres Bild jedes einzelnen Proteins, bilden sie einen Mittelwert aus so vielen Kopien wie möglich - ähnlich wie Fotografen, die dasselbe Foto mit unterschiedlichen Belichtungen aufnehmen, um sie später zu einem perfekt belichteten Bild zu kombinieren. Entscheidend ist, dass man die verschiedenen Proteine im Bild richtig identifiziert und lokalisiert, bevor man sie mittelt. „Wir können Hunderte von Tomogrammen pro Tag erstellen, aber uns fehlten bisher Werkzeuge, um die Moleküle darin vollständig zu identifizieren", sagt Rice.
Handverlesen
Bisher haben die Forschenden in den Tomogrammen nach Übereinstimmungen mit bereits bekannten Molekülstrukturen gesucht. Die dazu benutzten Algorithmen sind jedoch fehleranfällig. Eine weitere Option ist die Identifizierung von Molekülen per Hand, die zwar eine qualitativ hochwertige Auswahl gewährleistet, jedoch Tage bis Wochen pro Datensatz in Anspruch nimmt.
Weiterhin ist der Einsatz einer Form des überwachten maschinellen Lernens möglich. Diese Tools können sehr genau sein, sind aber derzeit nicht sehr benutzerfreundlich, da die Software für jedes neue Protein mit manueller Kennzeichnung von Tausenden von Beispielen trainiert werden muss - eine schier unmögliche Aufgabe für kleine biologische Moleküle in einer überfüllten zellulären Umgebung.
TomoTwin
Die neu entwickelte Software TomoTwin überwindet viele dieser Hürden: Sie lernt, die sich in ihrer Form innerhalb eines Tomogramms ähnelnden Moleküle herauszufiltern und ordnet sie einem geometrischen Raum zu - das System wird belohnt, wenn es ähnliche Proteine nahe beieinander platziert, und bestraft, wenn es das nicht tut. In der entstehenden Karte können die Forschenden die verschiedenen Proteine isolieren und genau identifizieren und sie auf diese Weise in der Zelle lokalisieren. "Ein Vorteil von TomoTwin ist, dass wir ein vortrainiertes „Picking-Modell“ anbieten", sagt Rice. Durch den Wegfall des Trainingsschritts kann die Software sogar auf lokalen Computern laufen - wo die Verarbeitung eines Tomogramms bisher 60-90 Minuten dauerte, reduziert sich die Laufzeit auf dem MPI-Supercomputer Raven auf 15 Minuten pro Tomogramm.
TomoTwin ermöglicht es den Forschenden, Dutzende von Tomogrammen in der Zeit auszuwählen, die sonst für die manuelle Auswahl eines einzigen Tomogramms erforderlich ist. Dies erhöht den Datendurchsatz aber auch die mittlere Geschwindigkeit ein besseres Bild zu erhalten. Derzeit kann die Software nur globuläre Proteine oder Proteinkomplexe mit einer Größe von mehr als 150 Kilodalton in Zellen aufspüren; in Zukunft will die Raunser-Gruppe auch Membranproteine, fadenförmige Proteine und Proteine mit geringerer Größe einbeziehen.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft



















Diese Produkte könnten Sie interessieren

Antibody Stabilizer von CANDOR Bioscience
Protein- und Antikörperstabilisierung leicht gemacht
Langzeitlagerung ohne Einfrieren – Einfache Anwendung, zuverlässiger Schutz

DynaPro NanoStar II von Wyatt Technology
NanoStar II: DLS und SLS mit Touch-Bedienung
Größe, Partikelkonzentration und mehr für Proteine, Viren und andere Biomoleküle

Meistgelesene News











Weitere News von unseren anderen Portalen
























Zuletzt betrachtete Inhalte
Schwarzer_Senf

Einzigartiges Nasenmikrobiom
