Verirrte Proteine lösen Erbkrankheiten aus
Die Krampfanfälle beginnen meist nur wenige Monate nach der Geburt. Doch die richtige Diagnose erhalten die Betroffenen mit dem seltenen Glut1-Defizit-Syndrom häufig erst nach Jahren. Unbehandelt entwickeln sich die erkrankten Kinder nur langsam und leiden häufig unter neurologischen Problemen. Dem Syndrom liegen verschiedene Defekte in einem Gen zugrunde. Durch sie verliert das Glut1-Protein in der Zellmembran seine Funktion: es befördert keinen Zucker mehr aus dem Blut ins Gehirn.
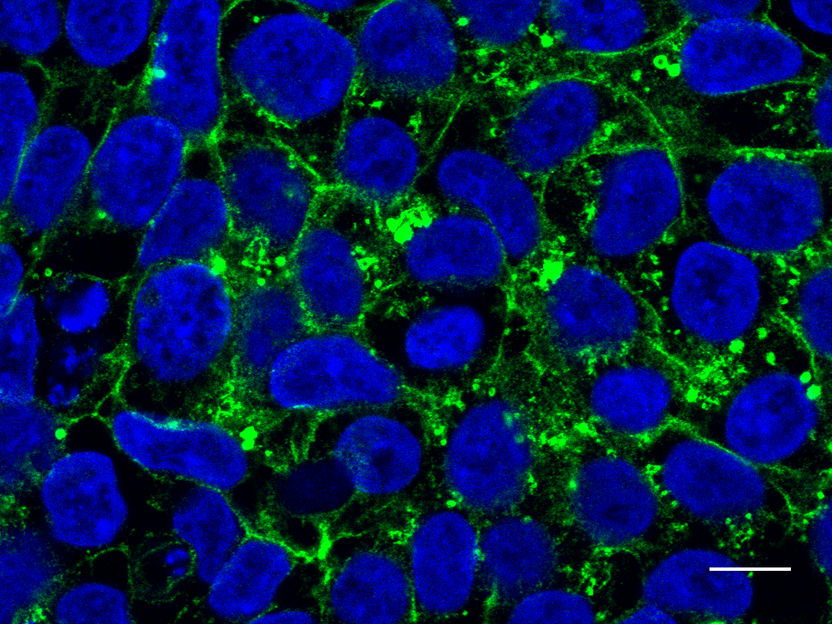
In den induzierten pluripotenten Stammzellen der Patientin befindet sich das Glut1-Protein vor allem im Zellinneren (grün markiert).
Katrina Meyer, MDC
Kleinste Veränderungen in bisher wenig beachteten, flexiblen Teilen des Glut1-Proteins können zu den weitreichenden Störungen in der Zelle führen. Weitere Erbkrankheiten könnten die gleiche Ursache haben. Dies zeigte das Forschungsteam um Professor Matthias Selbach vom Max-Delbrück-Centrum für Molekulare Medizin (MDC) in der aktuellen Ausgabe des Fachblattes Cell.
Ein grundlegendes Problem
Selbachs Team ging einer elementaren Frage nach: Wie verursachen defekte Gene Krankheiten? Im Glut1-Gen gibt es viele Stellen, an denen eine Mutation die dreidimensionale Gestalt des Glut1-Proteins zerstört, womit es seine Funktion verliert. Verformt und verbogen kann es seine Aufgabe in der Maschinerie der Zelle nicht mehr erfüllen und löst so das Syndrom aus. Genauso ist es im Prinzip auch bei anderen Erbkrankheiten. „Aber häufig ist der Mechanismus für eine genetisch bedingte Erkrankung, also die Ursache auf molekularer Ebene, unklar“, sagt Katrina Meyer, Doktorandin im Team von Selbach.
Bei einem Fünftel aller erblichen Erkrankungen scheine die Struktur des Proteins gar nicht beeinträchtigt zu sein, sagt die Wissenschaftlerin. Die Mutation trete in diesen Fällen an flexiblen Anhängseln der Proteine auf, von denen man bis vor kurzem noch dachte, dass sie keine Funktion haben, weil sie keine definierte Struktur besitzen. Doch der Anschein trügt: „Diese sogenannten intrinsisch ungeordneten Proteinregionen können sich wie ein Kissen an andere Proteine anschmiegen und sie so beeinflussen.“
Auf solchen Interaktionen zwischen Proteinen beruhen viele Vorgänge in der Zelle. Die Moleküle greifen in ineinander wie Zahnräder, übertragen Kräfte oder setzen Hebel und Förderbänder in Bewegung. Passt nun ein Protein nicht mehr an die gewohnte Stelle der Zellmaschine, kann das drastische Folgen haben. Meyer untersuchte deshalb zunächst, welche Proteine der Zelle mit mutierten und flexiblen Proteinregionen in Kontakt treten.
Unscheinbare Veränderung mit starken Auswirkungen
Die Doktorandin baute dafür 258 flexible Proteinregionen im Reagenzglas nach – „gesunde“ Varianten genauso wie krankheitsverursachende – und gab anschließend Extrakte aus menschlichen Zellen dazu. Mittels Massenspektrometrie prüfte die Wissenschaftlerin anschließend, welche Eiweiße mit den künstlichen Proteinen interagierten.
Meistens banden mutierte und „gesunde“ Regionen in Meyers Experiment an dieselben Bindungspartner. Doch ein Teil der mutierten Proteine verlor diese Fähigkeit ganz oder dockte an andere Eiweiße und störte so den Lauf der zellulären Maschinerie. Einige Genveränderungen beeinflussten auf diese Weise sogar den Transport von Proteinen innerhalb der Zelle. Darunter: eine Mutation im Gen für das Glut1-Protein.
Durch die Genveränderung lagen zwei Leucin-Proteinbausteine nebeneinander. „Dieses Muster lockt bekanntermaßen Proteine an, die für den Transport von Molekülen innerhalb der Zelle wichtig sind“, sagt Meyer.
In der Zelle verirrt
Als Meyer dieses Ergebnis in den Händen hielt, war das für sie ein besonderer Moment. Könnte es sein, dass bei Betroffenen mit dieser Mutation nicht das Glut1-Protein defekt ist, sondern es nur an der falschen Stelle in der Zelle landet? „Wenn nicht das Protein selbst betroffen ist, sondern nur sein Transport, gibt es die Chance, die zugrundeliegende Ursache zu behandeln und nicht nur das Symptom.“
Meyer durchstöberte Datenbanken und machte eine Patientin mit Glut1-Defizit ausfindig, bei der die Proteinregion zu einem Muster mit zwei Leucin-Bausteinen mutiert war. Von ihr bekam sie Zellen gespendet. Im Experiment mit den Zellkulturen zeigte Meyer, dass das mutierte Glut1 sich nicht mehr an der Außenseite der Zellen befand, wo das Protein den Zucker aufnimmt. Stattdessen befanden sie sich im Inneren der Zelle, als hätte es sich verirrt.
Verantwortlich für den Irrweg von Glut1 war unter anderem der zelluläre Apparat, der Bläschen der Zellmembran abschnürt und durch „Endozytose“ ins Zellinnere transportiert. Meyer konnte ihre These bestätigen: Blockierte sie diesen Prozess, gelangte das Glut1-Protein zurück an die Zellhülle und nahm dort wieder Zucker auf. „Das ließe sich theoretisch auch mit Medikamenten blockieren“, sagt Meyer.
Ein neuer Mechanismus für zahlreiche Krankheiten
Diese Medikamente existieren noch nicht, sagt Laborleiter Matthias Selbach. Doch die Entdeckung sei bei weitem nicht auf Glut1 beschränkt. In Datenbanken fand das Forschungsteam das Dileucin-Motiv elf Mal in den flexiblen Regionen von acht Proteinen, darunter jene, die die Stoffwechselkrankheit Mukoviszidose auslösen.
„Wir haben einen vielversprechenden Angriffspunkt gegen verschiedenste Krankheiten entdeckt“, sagt Selbach. „Ich sehe da ein gewisses Potential für neue Arzneimittel.“ Ob sich diese Erkrankungen systematisch, etwa mit Endozytose-Blockern, bekämpfen lassen, müssten zukünftige Studien nun erst zeigen.
Obwohl die Patienten mit Glut1-Defizit-Syndrom häufig jahrelang auf eine korrekte Diagnose warteten, sei die Krankheit heute relativ gut behandelbar, sagt Selbach. Doch für Betroffenen sind Nudeln, Brot oder ein Eis tabu. Sie müssen sich zucker- und stärkefrei ernähren, also eine streng ketogene Diät einhalten. Dann hören die Krampfanfälle häufig auf, weil sich die Gehirnzellen ihre Energie auf andere Weise holen. „Die Krankheit bleibt aber unheilbar und die Patienten müssen ihre Ernährung extrem einschränken.“
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft




















Diese Produkte könnten Sie interessieren

Antibody Stabilizer von CANDOR Bioscience
Protein- und Antikörperstabilisierung leicht gemacht
Langzeitlagerung ohne Einfrieren – Einfache Anwendung, zuverlässiger Schutz

DynaPro NanoStar II von Wyatt Technology
NanoStar II: DLS und SLS mit Touch-Bedienung
Größe, Partikelkonzentration und mehr für Proteine, Viren und andere Biomoleküle


































