Wissenschaftler verbessern Referenz-Genom des Chinesischen Hamsters
Weniger Lücken bei der Sequenzierung – Auswertung von enormen Datenmengen
Das Genom des Chinesischen Hamsters ist von Gießener Wissenschaftlern deutlich verbessert worden. In der Fachzeitschrift „Biotechnology and Bioengineering“ konnten sie in Zusammenarbeit mit einem internationalen Forschungsteam kürzlich ein Genom veröffentlichen, das eine ähnlich hohe Qualität aufweist wie die Genome der Modellorganismen Maus oder Ratte. Im Vergleich zu den ersten Versionen aus dem Jahr 2013 konnten in der verbesserten Version die Zahl der Genomfragmente deutlich verringert und viele Lücken geschlossen werden. Damit stehen der Wissenschaft jetzt mehr Informationen zu den vorhergesagten Genen und ihren regulatorischen Bereichen zur Verfügung. Die Herstellung neuer Proteine sowie die Optimierung bestehender Produktionsprozesse können somit noch zielgerichteter durchgeführt und weitaus besser kontrolliert werden.
In der Biotechnologie werden Zelllinien von unterschiedlichen Organismen zur Produktion von Medikamenten, Enzymen oder Chemikalien verwendet. Neben Bakterien oder Hefen kommen auch Zelllinien höherer Lebewesen wie Maus oder Mensch zum Einsatz. Eine der wichtigsten Zelllinien zur biotechnologischen Herstellung von Proteinen wurde vor über 50 Jahren in den USA von Theodore T. Puck aus Ovarien des Chinesischen Hamsters isoliert. Aus dieser ersten so genannten CHO-Zelllinie (engl.: Chinese Hamster Ovary cell line) wurden im Laufe der Zeit verschiedene, spezialisierte CHO-Zelllinien abgeleitet. Ihr schnelles Wachstum in Flüssigkulturen und ihre ertragreiche Produktion von Proteinen haben dazu beigetragen, dass CHO-Zelllinien heute in der Wissenschaft sowie in der pharmazeutischen Industrie verstärkt zum Einsatz kommen.
Ein weiterer Vorteil der CHO-Zelle ist, dass die mit ihrer Hilfe produzierten Proteine (zum Beispiel Antikörper) der biologischen Struktur von humanen Proteinen sehr ähnlich sind. Unter den diversen verfügbaren Säugetier-Zelllinien sind CHO-Zellen daher das am meisten verwendete Produktionsmedium für pharmazeutische Proteine. Ein wichtiges Werkzeug zur Optimierung des Produktionsprozesses und zur Erhöhung der Produktqualität ist die gentechnische Manipulation der verwendeten Zelllinien. Für diese Aufgabe ist die Entschlüsselung des Genoms eine wichtige Voraussetzung, denn mit der Genomsequenz lassen sich ganz gezielt und kontrolliert Veränderungen in das Erbgut der Zellen einbringen.
Entsprechend groß war das weltweite Interesse, das Genom des Chinesischen Hamsters zu entschlüsseln. Im Jahr 2013 wurde das Genom des Chinesischen Hamsters im Fachjournal Nature Biotechnology parallel von zwei Forscherteams erstmals publiziert. Neben einem Team aus den USA bestand das zweite Team aus deutschen und österreichischen Wissenschaftlerinnen und Wissenschaftlern, unter ihnen auch Prof. Dr. Alexander Goesmann von der Justus-Liebig-Universität Gießen (JLU).
Seither hat sich sowohl auf dem Sektor der Sequenziertechnologien als auch bei den technischen Möglichkeiten zur Auswertung der enormen Datenmengen einiges bewegt. Aus diesem Grunde haben sich die oben genannten Teams vor einiger Zeit zusammengeschlossen, um mit allen verfügbaren Daten die Qualität der Genomsequenz weiter zu verbessern. Für die erneute bioinformatische Auswertung wurde der Standort Gießen ausgewählt. Die Gruppe um Prof. Goesmann verfügt über eine jahrelange Erfahrung in der Entschlüsselung und Analyse von mikrobiellen Genomen und erstellte auch die erste Version des Hamstergenoms im Jahr 2013 für das deutsch-österreichische Team.
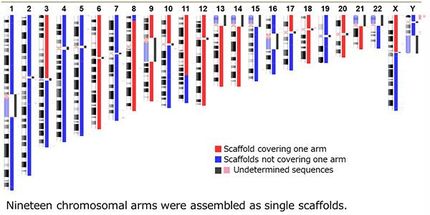
Das Genom des Chinesischen Hamsters hat mit elf Chromosomenpaaren zwar nur etwa halb so viele Chromosomen wie das menschliche Genom; mit über 2,3 Milliarden Basenpaaren ist es dennoch fast genau so groß und damit 500- bis 1000-mal größer als ein durchschnittliches Bakteriengenom. Für dieses Projekt wurden über zwei Milliarden kurze DNA-Sequenzen mit der Illumina Next-Generation-Sequencing-Methode erzeugt und zusätzlich fast 15 Millionen längere DNA-Sequenzen mit der Single-Molecule-Real-Time-Sequencing (SMRT)-Methode generiert.
Für die Verarbeitung der gesamten Rohdatenmenge von mehr als einem Terabyte mussten von den Gießener Bioinformatikerinnen und Bioinformatikern Prof. Goesmann, Dr. Karina Brinkrolf, Oliver Rupp und Sven Griep neue Methoden entwickelt und getestet werden. Allein die Berechnungen haben mehrere Monate gedauert; anschließend wurde noch einmal die gleiche Zeit benötigt, um die Ergebnisse zu bereinigen und zusammenzufassen. Hierfür standen ein Rechner-Pool mit über 1.000 CPU-Kernen und sehr leistungsfähige Einzel-Server mit bis zu zwei Terabyte Arbeitsspeicher zur Verfügung. Diese Hardwareausstattung wurde mitfinanziert vom Deutschen Netzwerk für Bioinformatik-Infrastruktur (de.NBI).
Originalveröffentlichung
Rupp O, MacDonald ML, Li S, Dhiman H, Polson S, Griep S, Heffner K, Hernandez I, Brinkrolf K, Jadhav V, Samoudi M, Hao H, Kingham B, Goesmann A, Betenbaugh MJ, Lewis NE, Borth N, Lee KH; "A reference genome of the Chinese hamster based on a hybrid assembly strategy"; Biotechnology and Bioengineering; 2018;1-14
Weitere News aus dem Ressort Wissenschaft





















Meistgelesene News











Weitere News von unseren anderen Portalen

























Zuletzt betrachtete Inhalte
Forschung im Bereich Biophotonik könnte wichtige Fortschritte in der Biotechnologie und Medizin bringen - Bewerbungsfrist für Kaiser-Friedrich Forschungspreis im Bereich Biophotonik läuft
CMC Biologics gibt Vertragsabschluss mit Daiichi Sankyo zur Entwicklung und Herstellung von Antikörpern bekannt
Novartis: A-Pandemie-Impfstoff Celtura erhält Zulassung in Deutschland
Flinke Enzyme mit zwei Fingern - Forscher der RUB und vom MPI Dortmund untersuchen kleine GTPasen

Bahrain gibt Corona-Impfstoff zum Notfallgebrauch für Helfer frei - Der Impfstoff stehe ab sofort zur Verfügung, seine Nutzung sei freiwillig
